
Since COVID-19’s emergence, our understanding of viruses has vastly increased and is driving novel innovations against older foes. New research examines the movement patterns of influenza surface proteins, which may aid in identifying potential weaknesses and improving future vaccines.

The early part of the 2020s has been dominated by COVID-19—but beyond its impacts on our health, the pandemic has driven incredible leaps forward in virology and vaccine technology. Now, alongside the lingering SARS-CoV-2 strains, influenza (flu) and respiratory syncytial virus (RSV) are causing concern for human health.
Where available, vaccines remain our strongest defense, but the flu vaccine has traditionally had to change each year to keep up with constant evolution and antigenic drift. This is because the glycoproteins on the flu virus that are used as the vaccine target can vary in number, position, and antigenicity from year to year.
This natural evolutionary process requires flu vaccines to be regularly updated to match the current strains—or those that are predicted to be predominant in the coming season. But we don’t always get it right, with variable vaccine effectiveness ranging from 10–60% according to CDC estimates from 2004–2022.1
Influenza is a single-stranded RNA virus that contains two glycoproteins: hemagglutinin and neuraminidase. And now, a study published in ACS Central Science sheds new light on how these proteins move and interact, revealing antigenically relevant states and new sites of vulnerability.1
It turns out that these glycoproteins are quite flexible, and they naturally move and “dance” in particular patterns. Previously, whole-virion protein dynamics have been challenging to study and difficult to interpret. But through multiscale computational methods, the researchers were able to create previously unseen views of the virus surface and better examine these proteins’ dynamics in situ.
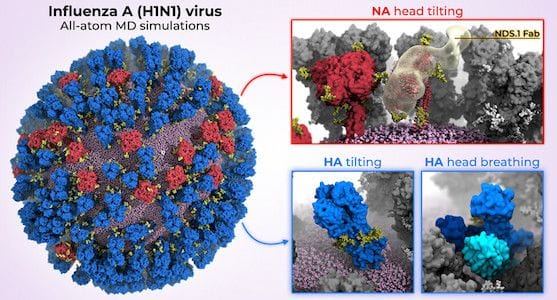
The fascinating results show that neuraminidases look like a pin, with a globular head on top of a thin stalk. Simulations show the head can tilt down more than 90 degrees.
The hemagglutinin protein also sticks up from the viral membrane, but it is connected by a flexible hinge. It can also tilt but not quite as much; however, it appears to regularly shift from a closed to a partially open position and back again.
Ultimately, the interplay and balance of these proteins could be the basis of viral fitness, since proteins usually do not form these sorts of clusters or interactions without a purpose.1,3 Importantly, this knowledge has a practical application: we can use it to gain access to the underside of the protein for broadly protective human antibodies.
These findings on how the flu surface proteins move reveals potential weaknesses that could be used to developed targeted drugs, and they also suggest that manipulating the glycoprotein position could be a way to develop more effective universal options against flu that would work regardless of the seasonal antigenic drift—giving us a better shot each year at achieving high vaccine effectiveness and protecting human health.
Watch the video around this research created by the ACS Science Communications team:
Explore related research in ACS Journals
All-Atom Mesoscale Simulations Predict the Conformational Dynamics of Influenza Virus Surface Glycoproteins
DOI: 10.1021/acscentsci.3c00020
Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism
DOI: 10.1021/acscentsci.9b01071
Influenza A M2 Inhibitor Binding Understood through Mechanisms of Excess Proton Stabilization and Channel Dynamics
DOI: 10.1021/jacs.0c06419
Proton-Induced Conformational and Hydration Dynamics in the Influenza A M2 Channel
DOI: 10.1021/jacs.9b05136
References
- Casolino L. et al. Breathing and Tilting: Mesoscale Simulations Illuminate Influenza Glycoprotein Vulnerabilities. ACS Cent. Sci. 2022, 8, 12, 1646–1663.
- Centers for Disease Control and Prevention. Past Seasons’ Vaccine Effectiveness Estimates. 2004-2022.
- Cohen, R.D. and Pielak, G.J. Electrostatic Contributions to Protein Quinary Structure. J. Am. Chem. Soc. 2016, 138, 40, 13139–13142.
